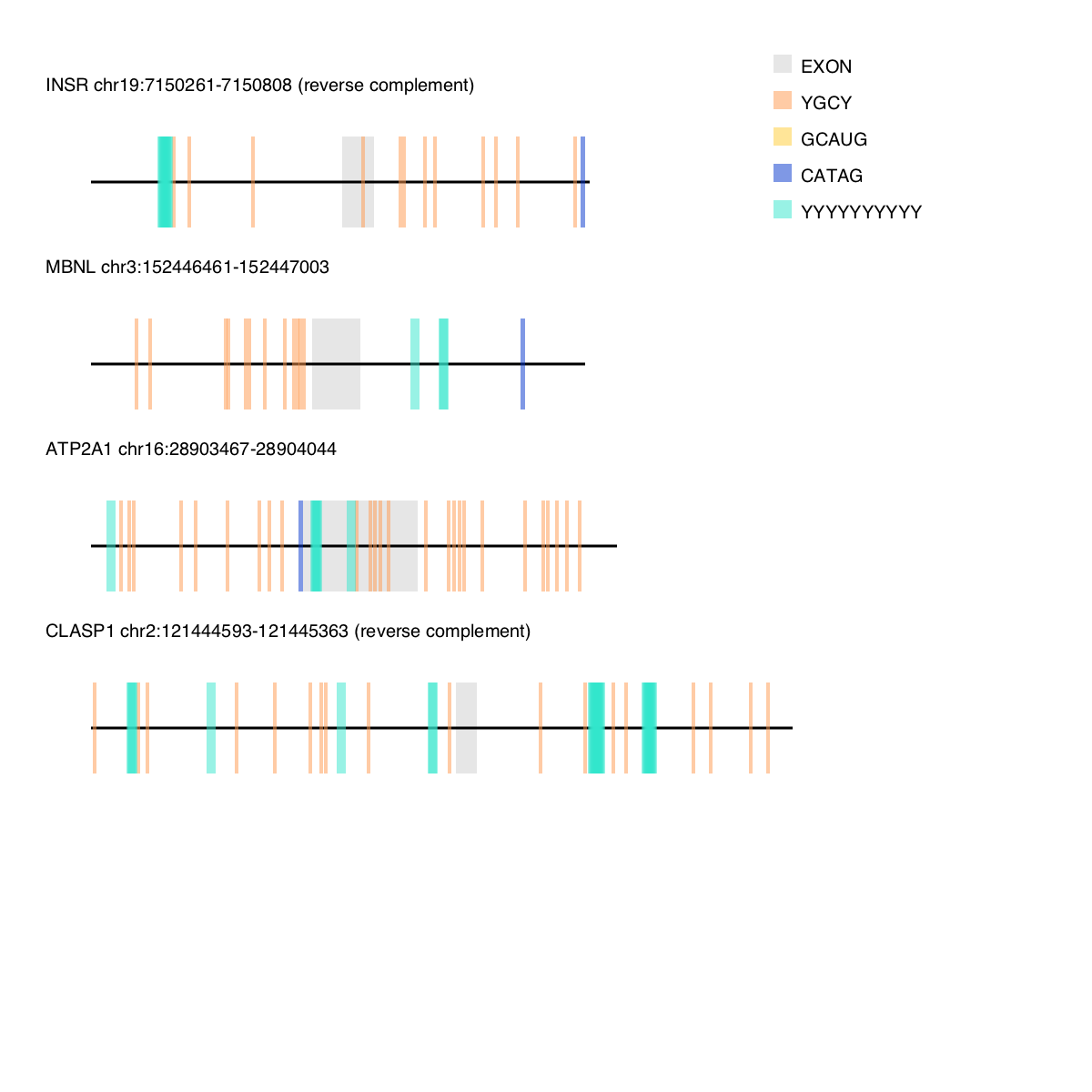
The script motif-mark-oop.py uses object oriented python code to visualize the locations of motifs on gene sequences.
- ambiguous nucleotide handling (Y,W,S,N etc).
- handles multipe gene sequences, and image size will increase with number of sequences.
- handles as many as 10 motifs.
- handles overlapping motifs - motifs are transparent
- visualizes exons and introns
The following modules must be installed in your environment prior to running motif-mark-oop.py
import itertools from itertools import product import re import cairo import argparse from IPython import display motif-mark-oop.py takes two input files: a file of motifs, and a fasta file. The motif file should have motifs separated by new lines. Example:
GCAUG ycgy YYYYYYYY CTAG The gene sequences should be in fasta format. NOTE: EXONS must be CAPITALIZED and introns must be lowercase. Example:
>MBNL chr3:152446461-152447003 tgtaattaactacaaagaggagttatcctcccaataacaactcagtagtgcctttattgt gcatgcttagtcttgttattcgttgtatatggcattccgatgatttgtttttttatttgt tttttctcacctacccaaaaatgcactgctgcccccatgatgcacctctgcttgctgttt atgttaatgcgcttgaaccccactggcccattgccatcatgtgctcgctgcctgctaatt aagACTCAGTCGGCTGTCAAATCACTGAAGCGACCCCTCGAGGCAACCTTTGACCTGgta ctatgacctttcaccttttagcttggcatgtagctttattgtagatacaagttttttttt taaatcaactttaaaatatatatccttttttctgttatagagttgtaaagtacaatgaaa Download motif-mark-oop.py from the Script folder in the same directory as your fasta file and motif file (otherwise, pass the complete path to your fasta file and motif file as arguments). The output png figure will be named with the same file prefix as the input fasta file, followed by ".png"
In the terminal, write:
./motif-mark-oop.py -f <fasta-file-name> -m <motif-file-name> motif-mark-oop.py will output a single png file with all of your sequences annotated like the example below: