Downloaded 39 times





![Breast cancer classification • Systematic investigation of dependency between expression patterns of thousands of genes (measured by DNA microarrays) and specific features of phenotypic variation in order to provide the basis for an improved taxonomy of cancer. • It is expected that variations in gene expression patterns in different tumors could provide a “molecular portrait” of each tumor, and that the tumors could be classified into subtypes based solely on the difference of expression patterns. • In litterature [20] classification techniques have been applied to identify a gene expression signature strongly predictive of a short interval to distant metastases in patients without tumor cells in local lymph nodes at diagnosis. In this context the number n of features equals the number of genes (ranging from 6000 to 30000) and the number N of samples is the number of patients under examinations (about hundreds). On the use of feature selection to deal with the curse of dimensionality in microarray data – p. 7/2](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-7-2048.jpg)

![Correlating motifs and expression levels • These methods consists in directly correlating expression levels and regulatory motif present in presumptive transcription control regions [4, 19]. • In [4], the expression of a gene in a single experimental condition is modelled as a linear function E = a1S1 + a2S2 + · · · + anSn of scores computed for sequence motifs in the upstream control region. These sequence motif scores incorporate the number of occurrences of the motifs and their positions with respect to the gene’s translation start site. • In other terms the sequence motifs of a specific gene are considered as explanatory variables (feature inputs) of a statistical model which correlates sequence features and expression of the gene. For instance the number of features is n = 4m for motifs of length m. On the use of feature selection to deal with the curse of dimensionality in microarray data – p. 9/2](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-9-2048.jpg)
![Feature selection • In recent years many applications of data mining (text mining, bioinformatics, sensor networks) deal with a very large number n of features (e.g. tens or hundreds of thousands of variables) and often comparably few samples. • In these cases, it is common practice to adopt feature selection algorithms [6] to improve the generalization accuracy. • There are many potential benefits of feature selection: • facilitating data visualization and data understanding, • reducing the measurement and storage requirements, • reducing training and utilization times, • defying the curse of dimensionality to improve prediction performance. On the use of feature selection to deal with the curse of dimensionality in microarray data – p. 10/2](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-10-2048.jpg)





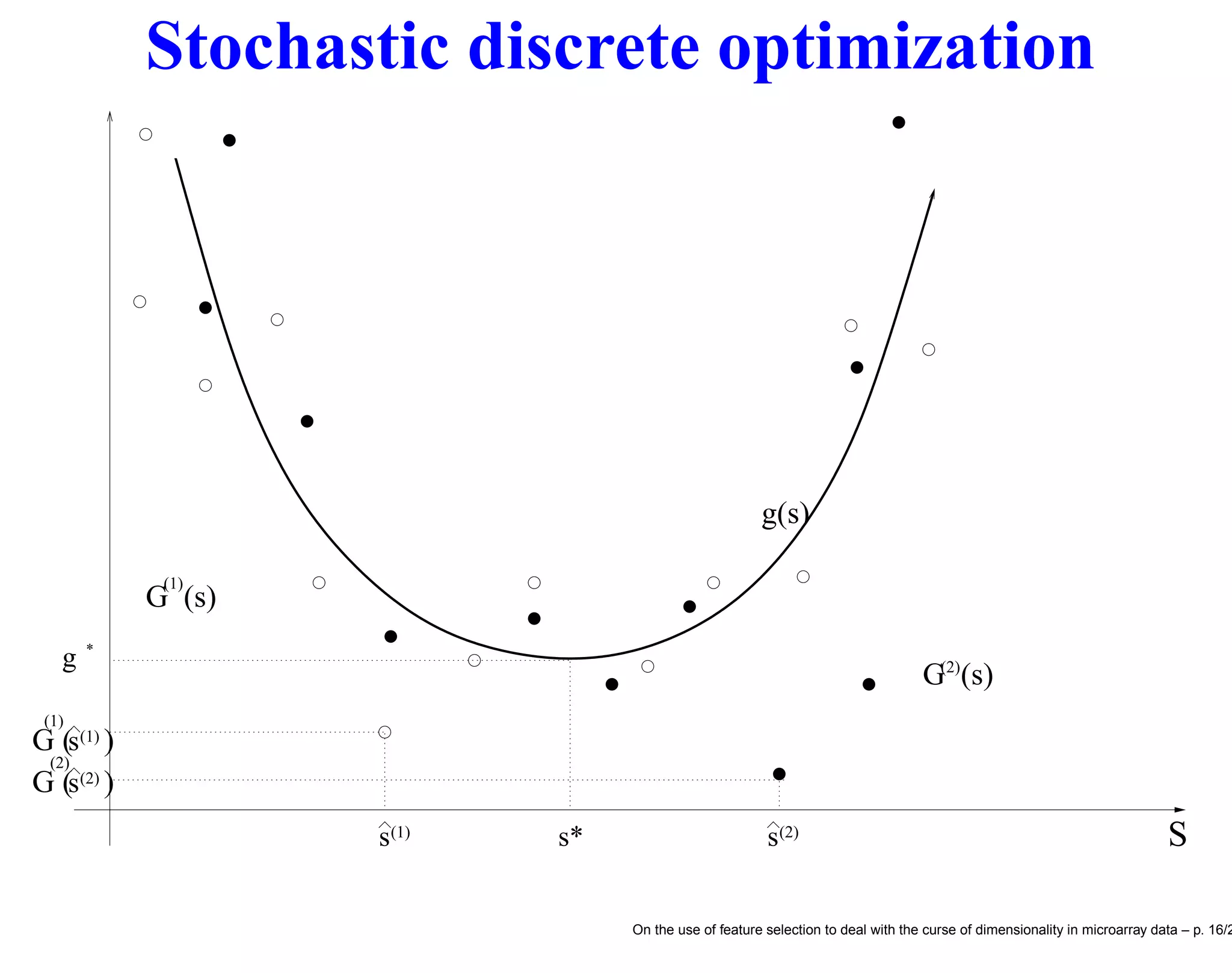
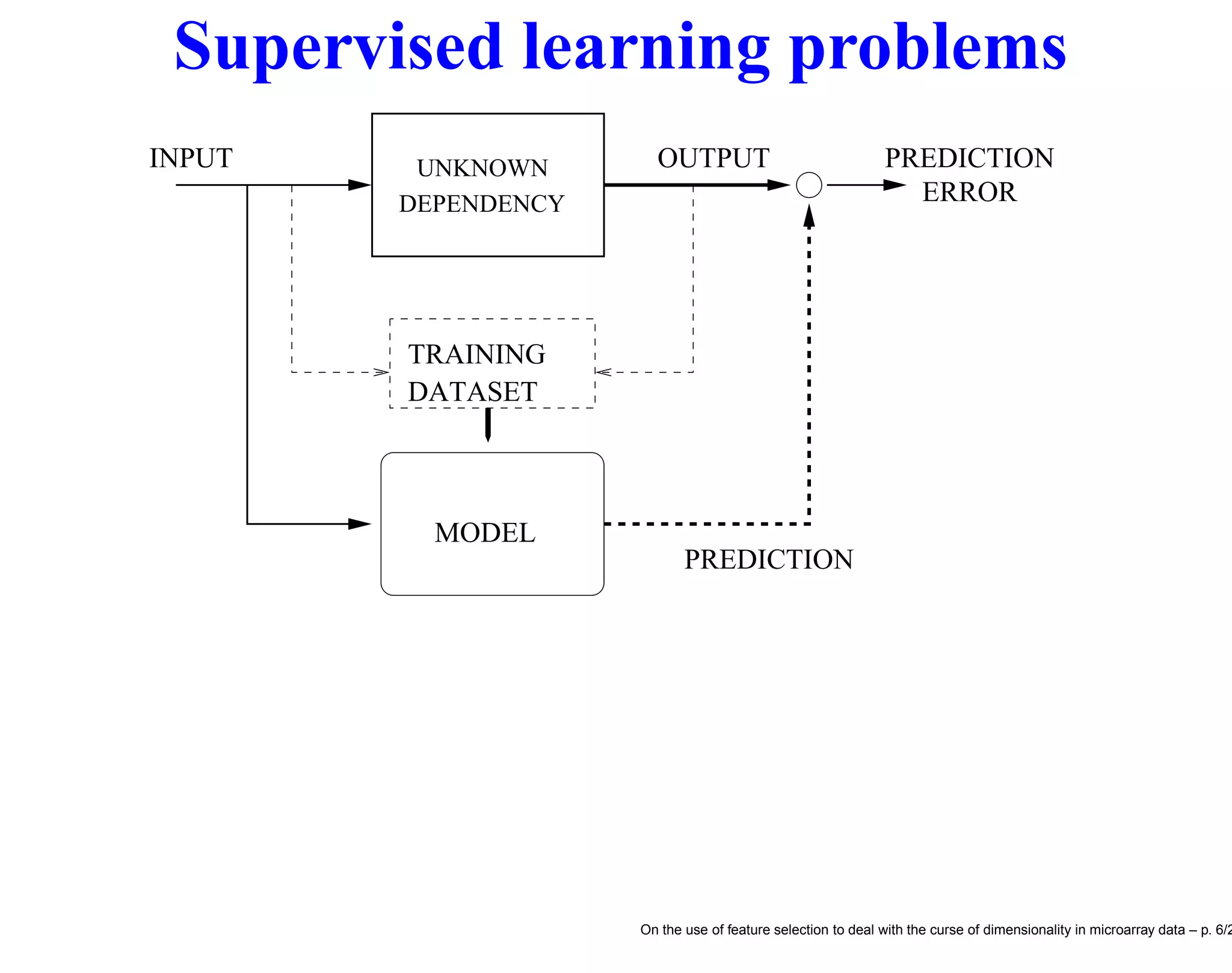
![F.s. and stochastic discrete optimization We may formulate the feature selection problem as a discrete optimization problem [10] s∗ = arg min s∈S Miscl(s) (1) be the optimal solution of the feature selection problem and the relative optimal generalization error, respectively. • Unfortunately the Miscl for a given s is not directly measurable but can only be estimated by the quantity Miscl(s) which is an unbiased estimator of Miscl(s). • The wrapper approach to feature selection aims to return the minimum ˆs of a cross-validation criterion Miscl(s) ˆs = arg min s∈S Miscl(s) (2) On the use of feature selection to deal with the curse of dimensionality in microarray data – p. 17/2](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-17-2048.jpg)
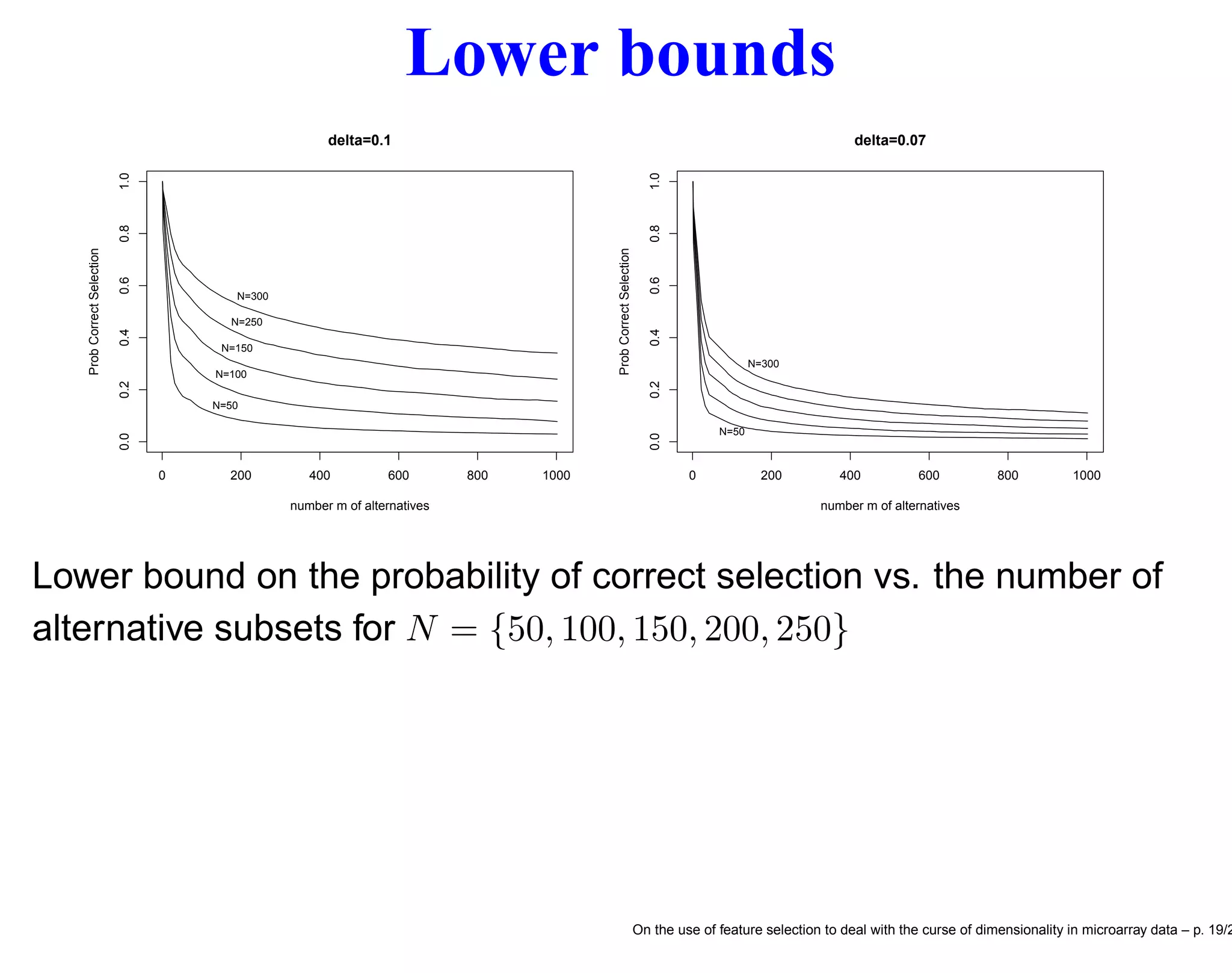
![Probability of correct selection Let Miscl(sj), j = 1, . . . , m be the (unknown) misclassification error of the jth feature subset and assume that the subsets are indexed in such a way that Miscl(sm) ≤ Miscl(sm−1) ≤ · · · ≤ Miscl(s1) so that (unknown to us) the feature set sm = s∗ is the best one. Assume also that Miscl(sm−1) − Miscl(sm) ≥ δ > 0. Prob {ˆs = s∗ } = Prob Misclm < Misclj, ∀j = m ≥ ≥ Prob Zj < δ σm 4/N , ∀j = m where Misclj − Misclm ≥ δ > 0 for all j = 1, . . . , m − 1 and the random vector (Z1, Z2, . . . , Zm−1) has a multivariate Normal distribution with means 0, variances 1 and common pairwise correlations 1/2 [9]. On the use of feature selection to deal with the curse of dimensionality in microarray data – p. 18/2](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-18-2048.jpg)

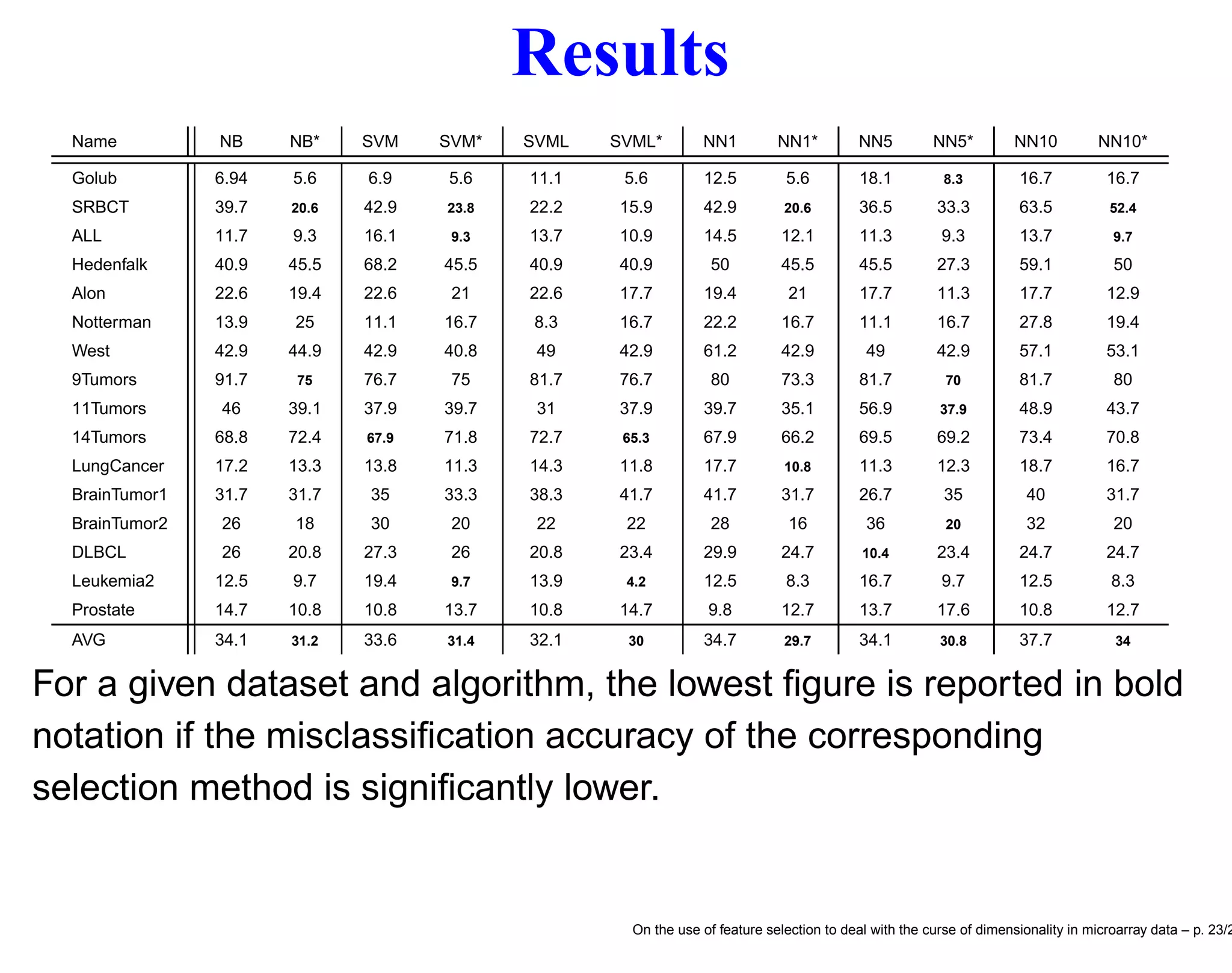
![Datasets Name Reference Platform N n C Golub [5] Affy hu6800 72 7129 2 SRBCT [8] cDNA 63 2308 4 ALL [22] Affy hgu95av2 248 12558 6 Hedenfalk [7] cDNA 22 3226 3 Alon [1] Affy hum6000 62 2000 2 Notterman [11] Affy hu6800 36 7457 2 West [21] Affy hu6800 49 7129 4 9Tumors [17] Affy hu6800 60 7129 9 11Tumors [18] Affy hgu95av2 174 12533 11 14Tumors [14] Affy hu35ksubacdf 308 15009 26 LungCancer [3] Affy hgu95av2 203 12600 5 BrainTumor1 [13] Affy hu6800 60 7129 5 BrainTumor2 [12] Affy hgu95av2 50 12625 4 DLBCL [15] Affy hu6800 77 7129 2 Leukemia2 [2] Affy hgu95a 72 12582 3 Prostate [16] Affy hgu95av2 102 12600 2 On the use of feature selection to deal with the curse of dimensionality in microarray data – p. 21/2](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-21-2048.jpg)



![References [1] U. Alon, N. Barkai, D.A. Notterman, K. Gish, S. Ybarra, D. Mack, and A.J. Levine. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc. of the National Academy of Sci- ences, 96(10):6745–6750, 1999. [2] S.A. Armstrong, J.E. Staunton, L.B. Silverman, R. Pieters, M.L. den Boer, M.D. Minden, S.E. Sallan, E.S. Lander, T.R. Golub, and S.J. Korsmeyer. Mll translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nature Genetics, 30(1):41–47, 2002. [3] A. Bhattacharjee, W.G. Richards, J. Staunton, C. Li, S. Monti, P. Vasa, C. Ladd, J. Beheshti, R. Bueno, M. Gillette, M. Loda, G. Weber, E.J. Mark, E.S. Lander, W. Wong, B.E. Johnson, T.R. Golub, D.J. Sugarbaker, and M. Meyerson. Classification of hu- man lung carcinomas by mrna expression profiling reveals dis- tinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A., 98(24):13790–13795, 2001. [4] H.J. Bussemaker, H. Li, and E. D. Siggia. Regulatory element detection using correlation with expression. Nature Genetics, 27:167–171, 2001. [5] T. R. Golub, D. K. Slonin, P. Tamayo, C. Huard, and M. Gaasen- beek. Molecular clssification of cancer: Class discovery and class prediction by gene expression monitoring. Science, 286:531–537, 1999. 25-1](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-26-2048.jpg)
![[6] I. Guyon and A. Elisseeff. An introduction to variable and feature selection. Journal of Machine Learning Research, 3:1157–1182, 2003. [7] I. Hedenfalk, D. Duggan, Y. Chen, M. Radmacher, M. Bittner, R. Simon, P. Meltzer, B. Gusterson, M. Esteller, O.P. Kallioniemi, B. Wilfond, A. Borg, and J. Trent. Gene expression profiles in hereditary breast cancer. New Engl. Jour. Medicine, 344(8):539– 548, 2001. [8] J. Khan, J. S. Wei, and M. Ringner. Clasification and diagnostic prediction of cancers using gene expression profiling and artifi- cial neural networks. Nature Medicine, 7(6):673–679, 2001. [9] S. H. Kim and B. L. Nelson. Handbooks in Operations Research and Management Science, chapter Selecting the Best System. Elsevier, 2005. [10] A. J. Kleywegt, A. Shapiro, and T. Homem de Mello. The sample average approximation method for stochastic discrete optimiza- tion. SIAM Journal of Optimization, 12:479–502, 2001. [11] D.A. Notterman, U. Alon, A.J. Sierk, and A.J. Levine. Transcrip- tional gene expression profiles of colorectal adenoma, adenocar- cinoma and normal tissue examined by oligonucleotide arrays. Cancer Research, 6:3124–3130, 2001. [12] C. L. Nutt, D.R. Mani, R.A. Betensky, P. Tamayo, J.G. Cairncross, C. Ladd, U. Pohl, C. Hartmann, M.E. McLaughlin, T.T. Batche- lor, P.M. Black, A. von Deimling, S.L. Pomeroy, T.R. Golub, and D.N. Louis. Gene expression-based classification of malignant 25-2](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-27-2048.jpg)
![gliomas correlates better with survival than histological classifi- cation. Cancer Research, 63(7):1602–1607, 2003. [13] S. L. Pomeroy, P. Tamayo, M. Gaasenbeek, L.M. Sturla, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis DN, Mesirov JP, Lander ES, and Golub TR. Prediction of cen- tral nervous system embryonal tumour outcome based on gene expression. Nature, 415(6870):436–442, 2002. [14] S. Ramaswamy, P. Tamayo, R. Rifkin, S. Mukherjee, C.H. Yeang, M. Angelo, C. Ladd, M. Reich, E. Latulippe, J.P. Mesirov, T. Pog- gio, W. Gerald, M. Loda, E.S. Lander, and T.R. Golub. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci U S A, 98(26):15149–15154, 2001. [15] M.A. Shipp, K.N. Ross, P. Tamayo, A.P. Weng, J.L. Kutok, R.C. Aguiar, M. Gaasenbeek, M. Angelo, M. Reich, G.S. Pinkus, T.S. Ray, M.A. Koval, K.W. Last, A. Norton, T.A. Lister, J. Mesirov andD.S. Neuberg, E.S. Lander, J.C. Aster, and T.R. Golub. Diffuse large b-cell lymphoma outcome prediction by gene- expression profiling and supervised machine learning. Nature Medicine, 8(1):68–74, 2002. [16] D. Singh, P.G. Febbo, K. Ross, D.G. Jackson, J. Manola, C. Ladd, P. Tamayo, A.A. Renshaw, A.V. D’Amico, J.P. Richie, E.S. Lander, M. Loda, P.W. Kantoff, T.R. Golub, and W.R. Sellers. Gene ex- pression correlates of clinical prostate cancer behavior. Cancer Cell., 1(2):203–209, 2002. 25-3](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-28-2048.jpg)
![[17] J.E. Staunton, D.K. Slonim, H.A. Coller, P. Tamayo, M.J Angelo, J. Park, U. Scherf, J.K. Lee, W.O. Reinhold, J.N. Weinstein, J.P. Mesirov, E.S. Lander, and T.R. Golub. Chemosensitivity pre- diction by transcriptional profiling. Proc Natl Acad Sci U S A., 98(19):10787–10792, 2001. [18] A.I. Su, J.B. Welsh, L.M. Sapinoso, S.G. Kern, P. Dimitrov, H. Lapp, P.G. Schultz, S.M. Powell, C.A. Moskaluk, H.F. Frier- son Jr, and G. M. Hampton G. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Re- search, 61(20):7388–7393, 2001. [19] S. L.M. van der Keles and M. B. Eisen. Identification of regula- tory elements using a feature selection method. Bioinformatics, 18:1167–1175, 2002. [20] L. J. van’t Veer, H. Dai, and M. J. van de Vijver. Gene expres- sion profiling predicts clinical outcome of breast cancer. Nature, 415:530–536, 2002. [21] M. West, C. Blanchette, H. Dressman, E. Huang, S. Ishida, R. Spang, H. Zuzan, J. R. Marks, and J. R. Nevins. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc. Nat. Acad. Sci., 98(20):11462–11467, 2001. [22] E.J. Yeoh, M.E. Ross, S.A. Shurtleff, W.K. Williams, D. Patel, R. Mahfouz, F.G. Behm, S.C. Raimondi, M.V. Relling, and A. Pa- tel. Classification, subtype discovery, and prediction of outcome in pediatric lymphoblastic leukemia by gene expression profiling. Cancer Cell, 1:133–143, 2002. 25-4](https://image.slidesharecdn.com/gand-160524075755/75/Feature-selection-and-microarray-data-29-2048.jpg)

This document discusses using feature selection techniques to address the curse of dimensionality in microarray data analysis. It presents the problem of having many more features than samples in bioinformatics tasks like cancer classification and network inference. It describes filter, wrapper and embedded feature selection approaches and proposes a blocking strategy that uses multiple learning algorithms to evaluate feature subsets in order to improve selection robustness when samples are limited. Finally, it lists several microarray gene expression datasets that are commonly used to evaluate feature selection methods.