Downloaded 61 times

















![ First: Calculate bit score S = Score of the alignment (Raw Score) , values depend on the scoring scheme and sequence composition of a database. [log value is natural logarithm (log base e)] BLAST Algorithm E-Value Calculation](https://image.slidesharecdn.com/presentationforblast-algorithmbio-informatice-160728161433/75/Presentation-for-blast-algorithm-bio-informatice-24-2048.jpg)





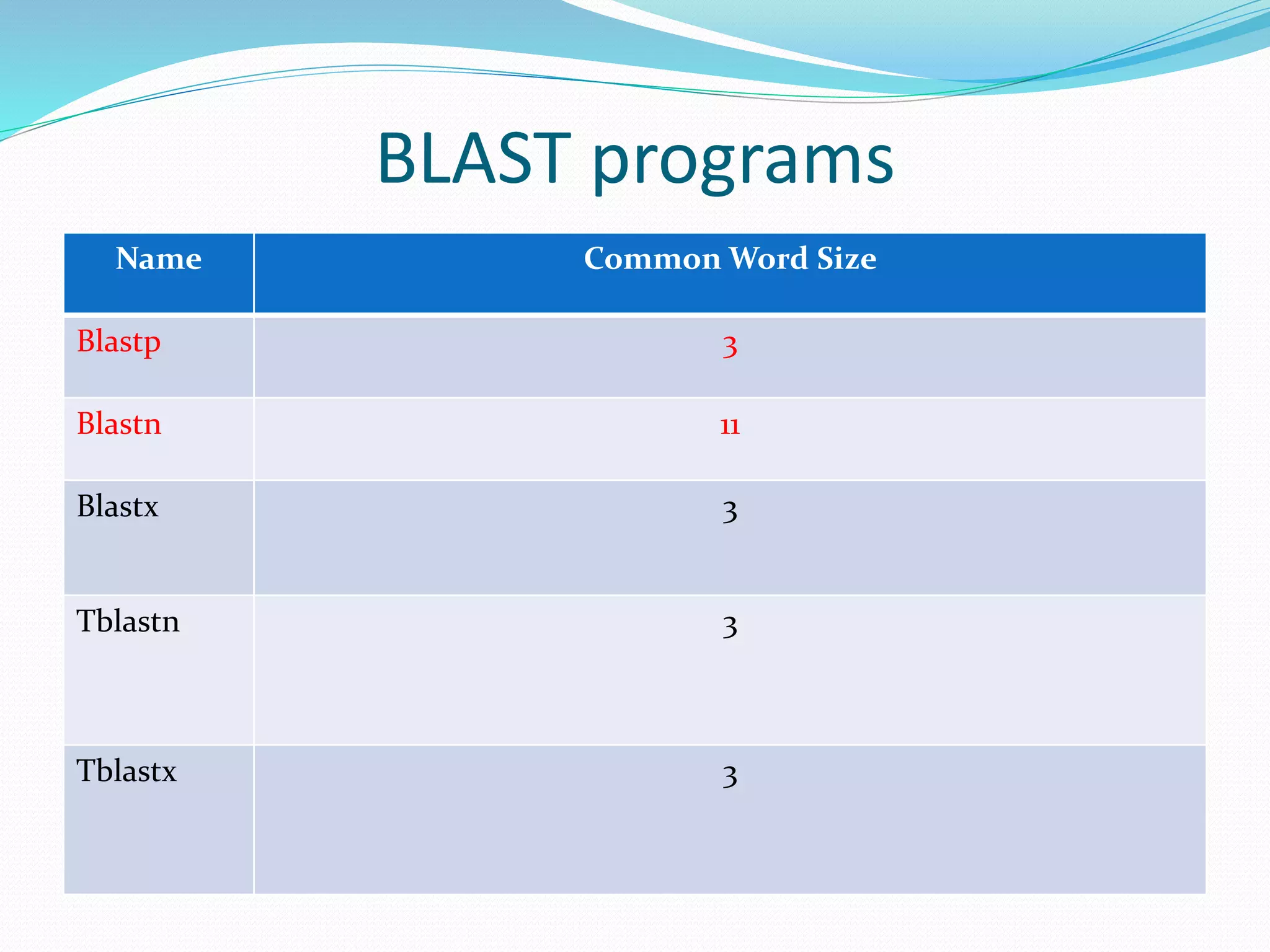



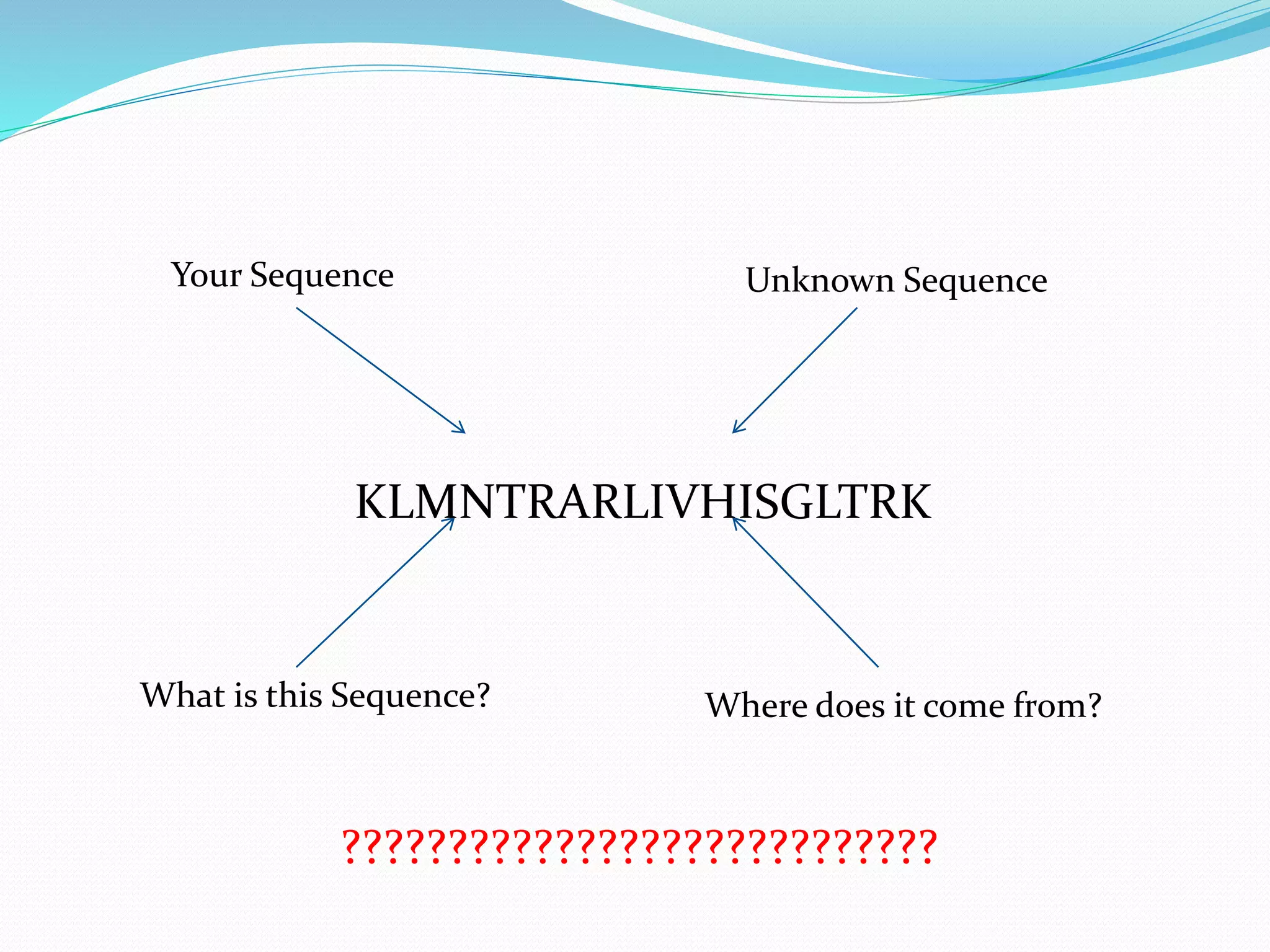
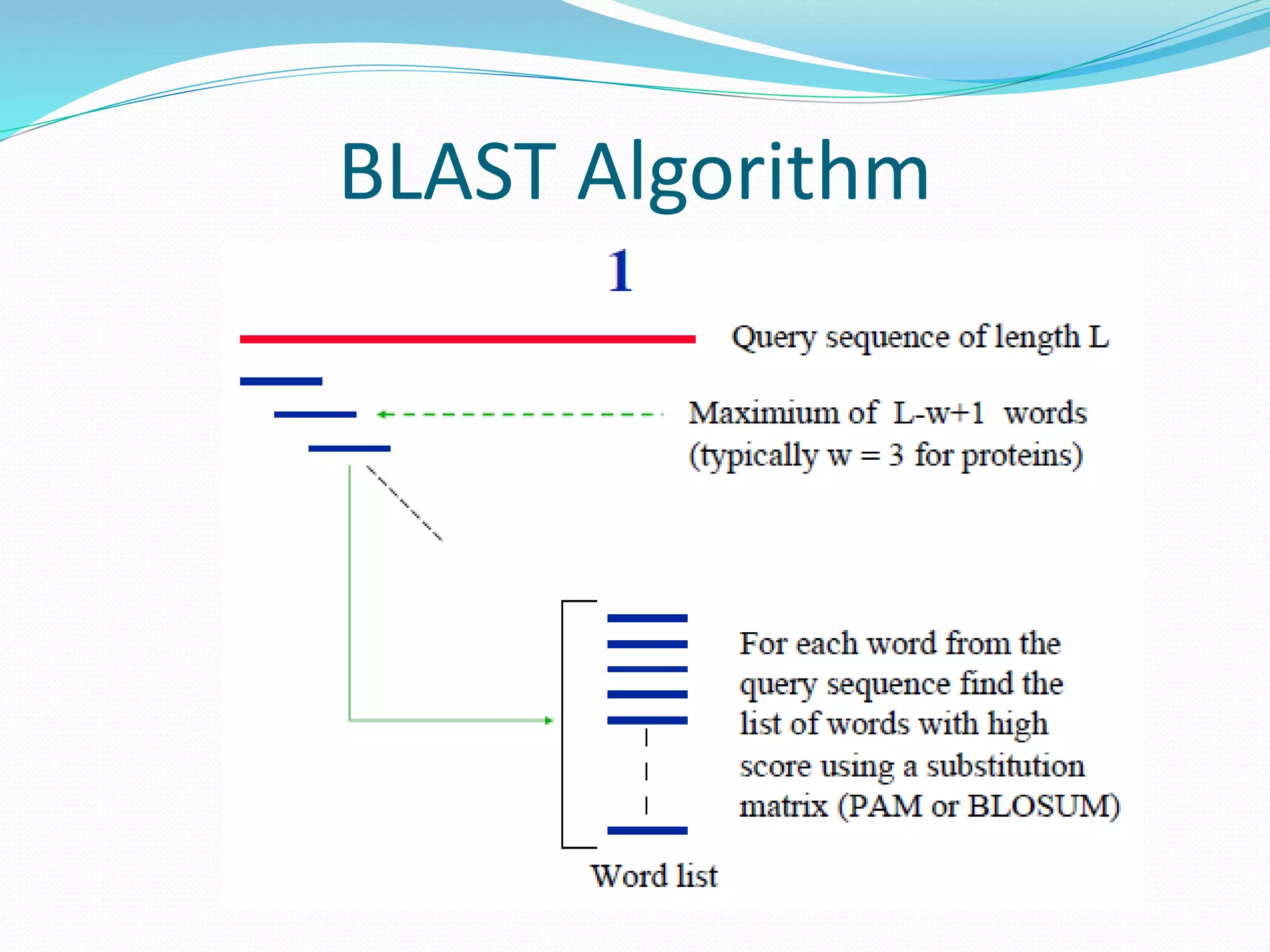
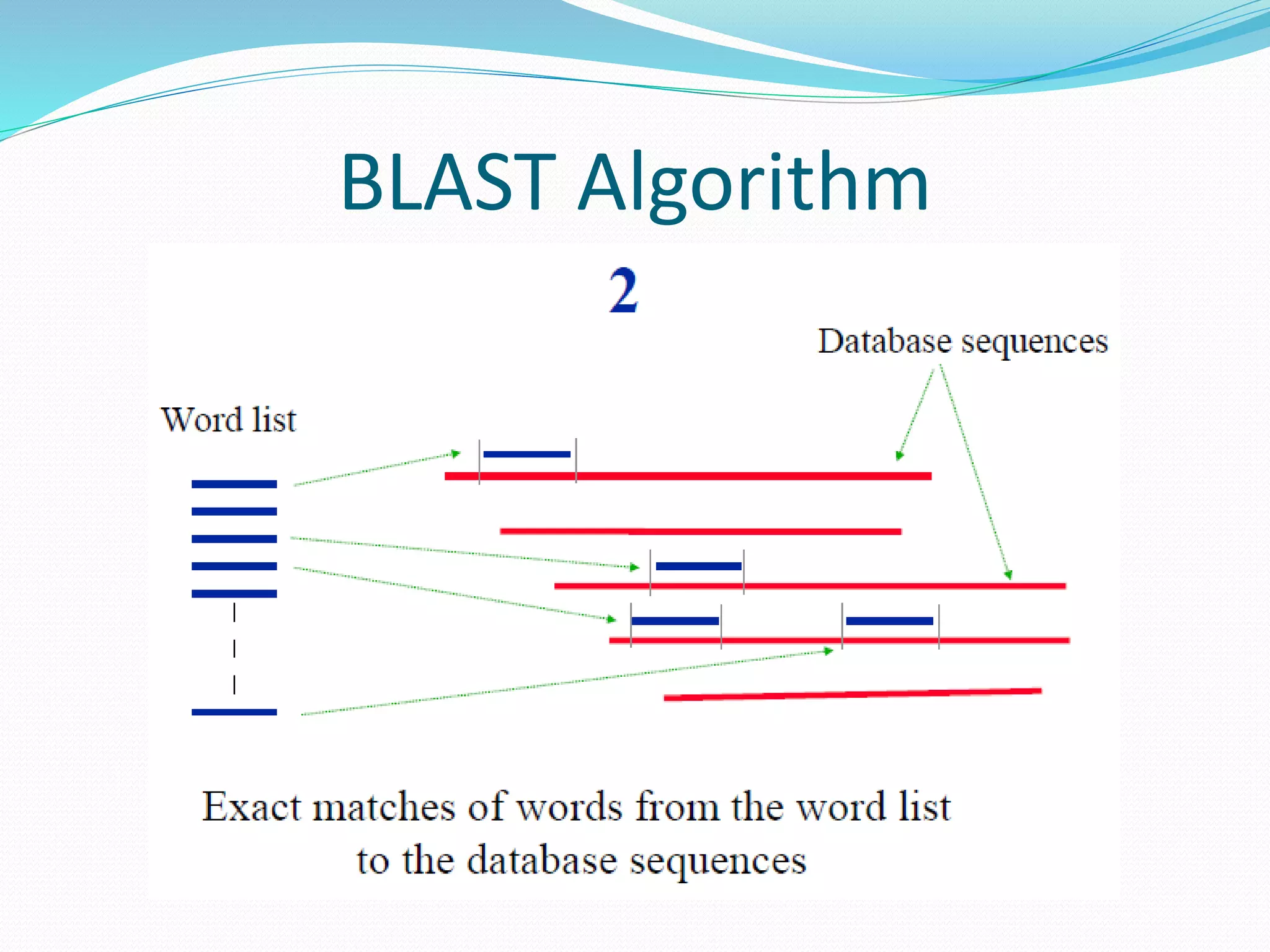
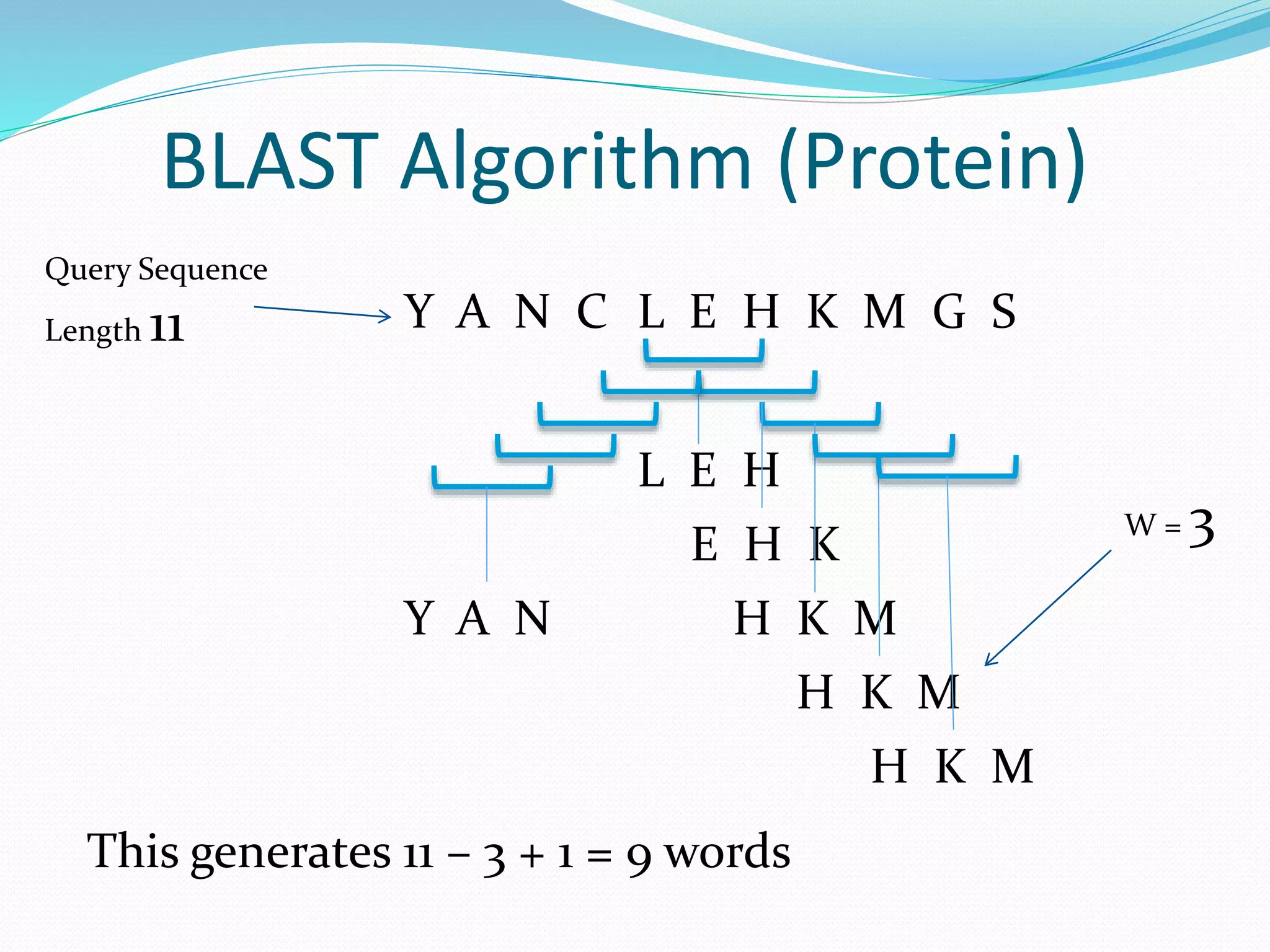
The document discusses the process of sequence alignment and the use of the Basic Local Alignment Search Tool (BLAST) to compare DNA/protein sequences with databases to infer structural and functional relationships. It explains the BLAST algorithm, its efficiency in database searching, and details about calculating scores and e-values to determine the significance of matches. Additionally, it provides information on various BLAST programs and recommendations for optimizing searches.